월요일, 1월 09, 2017
clustalw 설치
clustalw를 설치하기 위해서는 일단
다운로드를 받아야 하겠죠?
소스파일 clustalw2
프리컴파일 파일 clustalw2-libcppstatic
프리컴파일된 파일을 다운받아서 사용해도 되고
소스 파일을 다운받아서 설치하셔도 됩니다.
$tar zxf clustalw-2.1.tar.gz
$cd clustalw-2.1
$./configure --prefix=/install/path/clustalw-2.1/
$make install
그냥 이렇게 하시면 clustalw가 설치되고 잘 사용하시면되겠습니다. :)
목요일, 12월 29, 2016
SILVAngs
QIIME2이후 새로운 녀석이 나타났다!!
silvangs음.. 왠지
MG-RAST 대항마로 나온듯
MG-RAST야 분석 파이프라인만 제공하는것에 반해
silvangs의 경우 막강한 16S rRNA DB를 탑재하고 있으니...

그리고 silvangs도 일단은 credit으로 운영중 가입하면 300k주고 시작하는데
과금 방법이 분석횟수인지 분석에 필요한 스텝기준인지 아직
안사용해봐서 잘 모르겠다능.. :)
여튼 단점은 rDNA용 커뮤니티 분석이라는것이 한계
WGS가 플랫폼으로 나오면 대박일듯... 물론 WGS이 나오기에는 아직
넘어야할 산이 좀 있으니...
불안불안한 MG-RAST에 이어 web 기반 분석 플랫폼이 나와준것에 박수를..
목요일, 12월 15, 2016
HaploMerger2
HaploMerger2
HaploMerger2 논문
HaploMerger2 다운로드 사이트 , HM2 메뉴얼
2012년 HaploMerger1 개발 이후 2014년에 성능이 향상된 HaploMerger2로 2015년 11월자 파일이 최신인 프로그램으로 간단히 정리하면 polymorphism으로 인해 assembly가 제대로 되지 않는 genome을 assembly하여 고 퀄러티의 genome을 얻는데 도움을 주는 도구 되겠습니다. :)
기본적인 작업 프로세스는 아래의 순서도를 따릅니다.

HM2에서 제공되는 batch 스크립트를 보시면 위에 모식도를 그대로 구현해 놓은것이라고 보시면 됩니다.
HM2 설치는 다른 것들과 달리 컴파일하는거 없고 그냥 다운받아서 압축 파일만 풀면 되는데 run_all.batch파일이 속을 썩일수도 있습니다.
run_all.batch파일안에 치환해야 하는 파일 이름들 중 mp_ref/alt_ss이나 mp_ref/alt_ss_re이 것을
mp_ref_ss와 mp_alt_ss로 분리해주시면 중간에 에러나서 실행되지 않는 step들이 해결될것입니다. 그리고 추가적으로 우리 kent옹께서 만드신 faToNib도 필요하고 여기저기 숨어있는 풀어야할 문제들이 약간씩은 있을 수 있으나 log파일 확인하시면 다 나오는것이니 너무 걱정하지 마시고 일단 run 해보시면됩니다. libraries안에 gapCloser.cfg와 sspace_libraries.list는 GapCloser와 SSPACE할때 필요한 라이브러리 설정 파일입니다. 그 안에 형식에 맞게 fastq파일 넣어주시면됩니다.
마지막으로 HM2가 많이 궁금하신분은 제가 아는 선에는 HM2를 가장 잘 다루는 사람은 (주)테라젠이텍스의 박신기대리님이신듯!! 궁금한것 박대리님께!! 물어보십시요!! ㅋ :)
화요일, 12월 06, 2016
HGAP 버전 비교
PacBio에서 assembly를 담당하고 있는 HGAP과 Falcon을 주로 사용하고 있는것으로 알고 있는데 오늘 그 중에서 HGAP을 잠시 확인 하도록 하겠습니다.
왜 HGAP만 확인하느냐?
제가 HGAP을 많이 쓸 예정이거든요 ㅋ
Falcon궁금하시면 직접 하시면됩니다!! :)
우선 HGAP 버전을 한번 살펴보겠습니다.

HGAP버전을 보시면 3개가 존재합니다. 그러나 HGAPv1의 경우 이미 이번 SMRT버전에서 퇴출당했습니다. 당시에는 long read로 어셈블리해서 획기적으로 계산량을 줄이긴 했으나 Celera Assembler 자체가 시간이 많이 걸리기 때문에 (CA자체라기 보다는 특정 모듈들이겠죠?) 자체 프로그램을 고안해서 HGAPv2외에 추가로 HGAPv3까지 내놓은 상황입니다.
근데 보통 New버전 나오면 예전거 갈아치우는데(한국이 그런 경향이 많긴하지만.. 모 특정 위치에 있는 분들은 상당히 잘 안바뀌긴하죠..) HGAPv2와 HGAPv3를 계속 사용하고 있습니다. 두개의 큰차이는 consensus 부분인데 이것에 따라 속도와 결과물 차이가 나기때문에 두개 결과중에 좋은거 사용해라하는 느낌이 있습니다.
그래서 현재 HGAPv2와 HGAPv3에 대해서 테스트를 계획중이고
각 단계별로 parameter를 바꿔주면 어떻게 바뀌는지 비교해볼 계획입니다.
PacBio를 사용하시는 업자분들께서는 어떤 버전을 최적화해서 사용하는지는 잘 모르겠으나 아마 각각의 장단점이 있을겁니다. 그래서 직접 테스트를 해보겠다능~ :)
그럼 HGAP 비교글은 내년에 좀 더 볼만한 내용을 찾아뵙도록 하겠습니다. :)
getopts.pl 이 없다면!!!
perl script를 실행시킬때 Can't locate getopts.pl 이라는 에러가 났을때
당신의 OS가 우분투라면?
당신의 OS가 우분투라면?
apt-get install libperl4-corelibs-perl로 해결가능합니다. orz
월요일, 12월 05, 2016
16S rRNA와 시퀀싱플랫폼
블로그에 쓰는 내용이 16S rRNA에 많이 집중되고 있긴하죠? ㅎㅎ
하는일이 이거다보니.. :)
여튼 오늘은 16S rRNA와 시퀀싱플랫폼에 대해서 잠시 이야기 하도록 하겠습니다.
최근 16S rRNA sequencing의 최강자 454가 서비스를 bye bye한 관계로
많은 연구자들이 MiSeq체제로 변환하고 있는데 (물론 Ion도 있고, PacBio도 있습니다.)
기존에 454를 사용했을 때와 다른 V region을 사용하고 다소 다른 결과들을 보이는 것들이
있을 것 입니다. 그래서 이리저리 검색하다가 걸린 논문 두 개를 가지고 잠시 얘기해보고자 합니다.
Nucl. Acids Res. (2010) 38 (22): e200.
PeerJ (2016) 4:e1869
논문을 찾아보기 시작한 이유는 454가 막을 내린 후 다른 시퀀싱 플랫폼에서는 왜 다른 region을 target하고 있고 왜 diversity에 차이를 보이는지... (내가 분석을 잘못했나.. ㅎㄷㄷㄷ)
우선 V region, 454는 V1-V2였는데 MiSeq은 V4 region을, Ion은 4개 region? 7개 region? 을 동시에target하고 있다는..
모 논문보면 아시겠지만
2010년 논문은 454 vs MiSeq을 비교했습니다. 두 플랫폼이 차이가 날까? 어떤것이 차이가 날까?
2010년도 논문을 한장 figure 요약하자면 이거죠

상단은 phylum abundance/ 하단은 genus abundance 그리고 좌측에 있는 V4는 이전 연구에서 사용되었던 기준이라고 생각되는 참고용 분포입니다. 실험 결과가 V4 region이 저 분포를 나타내면 실험이 잘됐다고 확인하는 용도로 사용됩니다.
사실 phylum은 크게 차이없죠, 차이가 있기도 쉽지않습니다.
관심사는 genus되겠습니다. 일루미나 데이터의 대부분은 unclassified입니다. 논문에서는 error때문에 이렇게 나왔으니 error좀 낮아지면 일루미나가 output이 많으니 sequence error 문제점을 개선된다면 미생물 분석에 적합한 킹왕짱 시퀀서가 될거라고 하는데 개인적인 생각으로는 sequence error(454가 일루미나가 한테 sequence error ㅋㅋ 좀 웃겼다능..
systematic error는 눈에 안뵈냐라고 한다면 눼눼, 하긴 이 논문이 2010년이란 것을 감안한다면 무리는 아니긴 합니다.)보다는 db선택이 unclassified문제는 보정할 수 있지 않을까 합니다.
그렇다면 이제 16s rRNA 입문한지 얼마안된 님께서 그렇게 느끼는 느낌적인 이유는
무엇인가?
바로 2016년 논문되겠습니다.
2016년 논문은 PacBio로 시퀀싱한 것을 db에 따라 분석 결과가 달라지는냐에 대한 내용으로
다음 한 장으로 요약 할 수 있겠습니다.
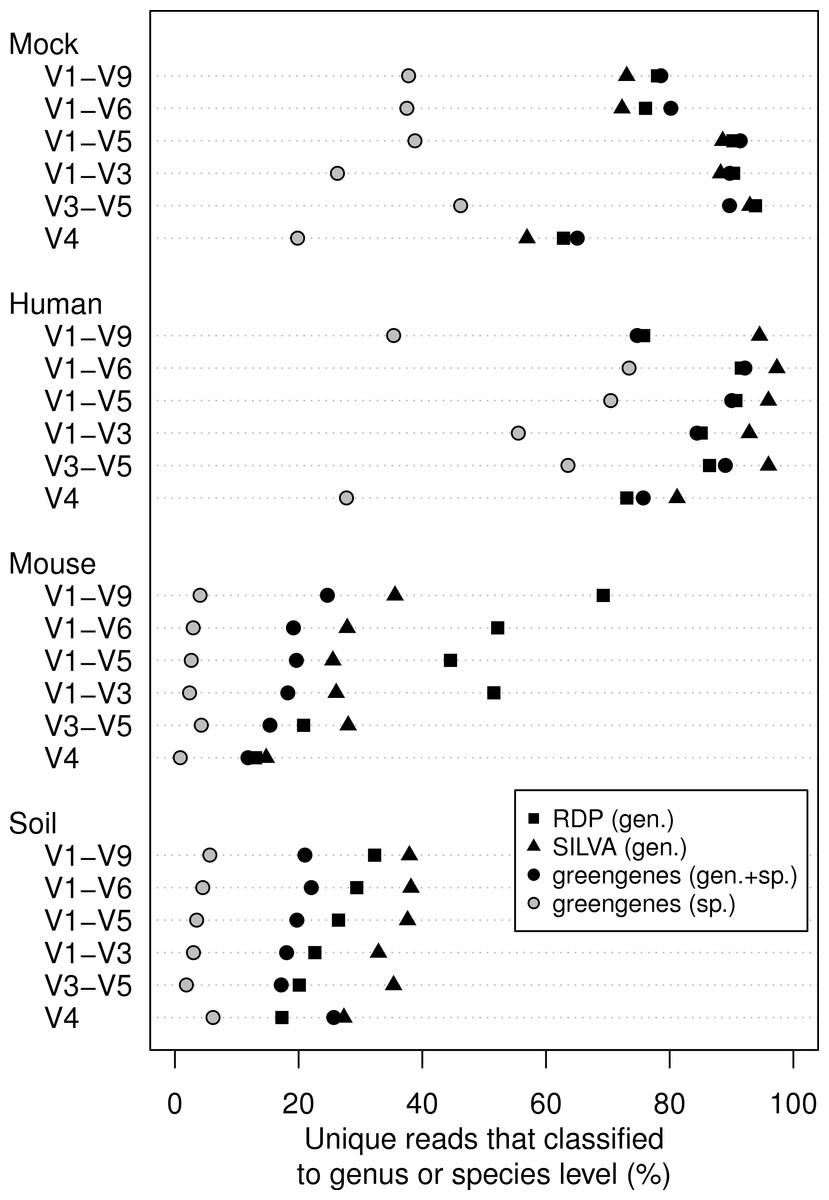
동일한 시료를 가지고 시퀀싱한 데이터를 16S rRNA db에 따라 classification되는 정도를 확인해본 그래프 입니다. 느낌 딹오시죠?
다만 나중에 뒤통수 맞았다는 느낌 안 받게 하나 말씀드리자면 RDP와 Silva의 경우 genus까지만 확인했고 gg는 genus와 genus+species 두가지로 확인한 것 입니다.
이제 PacBio의 Sequel에서 CCS로 생산된 16S rRNA 서열가지고 연구해도 나쁘지 않을 것 같다는..
아.. 이제 돈만 많으면 되는건가!!!
하는일이 이거다보니.. :)
여튼 오늘은 16S rRNA와 시퀀싱플랫폼에 대해서 잠시 이야기 하도록 하겠습니다.
최근 16S rRNA sequencing의 최강자 454가 서비스를 bye bye한 관계로
많은 연구자들이 MiSeq체제로 변환하고 있는데 (물론 Ion도 있고, PacBio도 있습니다.)
기존에 454를 사용했을 때와 다른 V region을 사용하고 다소 다른 결과들을 보이는 것들이
있을 것 입니다. 그래서 이리저리 검색하다가 걸린 논문 두 개를 가지고 잠시 얘기해보고자 합니다.
Nucl. Acids Res. (2010) 38 (22): e200.
PeerJ (2016) 4:e1869
논문을 찾아보기 시작한 이유는 454가 막을 내린 후 다른 시퀀싱 플랫폼에서는 왜 다른 region을 target하고 있고 왜 diversity에 차이를 보이는지... (내가 분석을 잘못했나.. ㅎㄷㄷㄷ)
우선 V region, 454는 V1-V2였는데 MiSeq은 V4 region을, Ion은 4개 region? 7개 region? 을 동시에target하고 있다는..
모 논문보면 아시겠지만
2010년 논문은 454 vs MiSeq을 비교했습니다. 두 플랫폼이 차이가 날까? 어떤것이 차이가 날까?
2010년도 논문을 한장 figure 요약하자면 이거죠

상단은 phylum abundance/ 하단은 genus abundance 그리고 좌측에 있는 V4는 이전 연구에서 사용되었던 기준이라고 생각되는 참고용 분포입니다. 실험 결과가 V4 region이 저 분포를 나타내면 실험이 잘됐다고 확인하는 용도로 사용됩니다.
사실 phylum은 크게 차이없죠, 차이가 있기도 쉽지않습니다.
관심사는 genus되겠습니다. 일루미나 데이터의 대부분은 unclassified입니다. 논문에서는 error때문에 이렇게 나왔으니 error좀 낮아지면 일루미나가 output이 많으니 sequence error 문제점을 개선된다면 미생물 분석에 적합한 킹왕짱 시퀀서가 될거라고 하는데 개인적인 생각으로는 sequence error(454가 일루미나가 한테 sequence error ㅋㅋ 좀 웃겼다능..
systematic error는 눈에 안뵈냐라고 한다면 눼눼, 하긴 이 논문이 2010년이란 것을 감안한다면 무리는 아니긴 합니다.)보다는 db선택이 unclassified문제는 보정할 수 있지 않을까 합니다.
그렇다면 이제 16s rRNA 입문한지 얼마안된 님께서 그렇게 느끼는 느낌적인 이유는
무엇인가?
바로 2016년 논문되겠습니다.
2016년 논문은 PacBio로 시퀀싱한 것을 db에 따라 분석 결과가 달라지는냐에 대한 내용으로
다음 한 장으로 요약 할 수 있겠습니다.
동일한 시료를 가지고 시퀀싱한 데이터를 16S rRNA db에 따라 classification되는 정도를 확인해본 그래프 입니다. 느낌 딹오시죠?
다만 나중에 뒤통수 맞았다는 느낌 안 받게 하나 말씀드리자면 RDP와 Silva의 경우 genus까지만 확인했고 gg는 genus와 genus+species 두가지로 확인한 것 입니다.
이제 PacBio의 Sequel에서 CCS로 생산된 16S rRNA 서열가지고 연구해도 나쁘지 않을 것 같다는..
아.. 이제 돈만 많으면 되는건가!!!
수요일, 11월 30, 2016
MrBayes 설치
MCMC방법으로 Phylogeny Tree를 그려주는 MrBayes!!
(컴파일해서 나오는 실행 파일명이 그닥 맘에 들지 않는 tool중 하나 ㅋㅋ)
자세한건 물어보시지 마시고 요기서는 설치만!
(그렇다고 다음에는 자세한거 말씀드리지는 않는다능)
자 설치를 위해서는 요기에서 말하는 Library는 미리미리 설치하면 암에 안 걸리니 먼저 잘 설치해주시기 바랍니다.
CentOS
Ubuntu
그리고 추가적으로 beagle를 설치하셔야 할겁니다.
Beagle 설치방법은 요기를 참고하시고 대략 밑에처럼 하시면 됩니다.
자세한건 물어보시지 마시고 요기서는 설치만!
(그렇다고 다음에는 자세한거 말씀드리지는 않는다능)
자 설치를 위해서는 요기에서 말하는 Library는 미리미리 설치하면 암에 안 걸리니 먼저 잘 설치해주시기 바랍니다.
CentOS
yum install make,automake,gcc,gcc-c++,kernel-devel,git,autoconf,automake,libtool,subversion,pkgconfig,java-1.6.0-openjdk-devel,openmpi,openmpi-develCentOS에서 Autoconf 버전으로 인해 문제가 발생했을 때는 그냥 컴파일 하세요. :)
$ wget http://ftp.gnu.org/gnu/autoconf/autoconf-2.69.tar.gz
$ tar xvfvz autoconf-2.69.tar.gz
$ cd autoconf-2.69
$ ./configure
$ make
$ sudo make install
Ubuntu
apt-get install build-essential,autoconf,automake,libtool,subversion,pkg-config,openjdk-6-jdk,git, openmpi-bin,openmpi-doc,libopenmpi-dev
그리고 추가적으로 beagle를 설치하셔야 할겁니다.
Beagle 설치방법은 요기를 참고하시고 대략 밑에처럼 하시면 됩니다.
git clone --depth=1 https://github.com/beagle-dev/beagle-lib.git
cd beagle-lib
./autogen.sh
./configure --prefix=$HOME
make install
마지막으로
MrBayes 소스파일을 다운로드 받으시고 설치해 주시면 되겠습니다.
MrBayes 소스파일을 다운로드 받으시고 설치해 주시면 되겠습니다.
$ autoconf
$ ./configure --prefix=/install/to/path --enable-mpi=yes
혹은
$ ./configure --prefix=/install/to/path --enable-mpi=yes --with-beagle=/install/to/path
$ make
화요일, 11월 29, 2016
QIIME2
QIIME2 출시(된지는 한달여정도... 지났지만..)
설치만 해두려고 qiime2 설치하다가 환경 설정이 꼬였는지
쓰고있던 qiime 1이 작동안해서 멘붕이 왔다는건 비밀;;; 빼고는
아직까지는 호감인 녀석!
여튼 이번 qiime2 출시하면서 변화를 준게 완전히 qiime 플랫폼을 만든듯..
통상적으로 사용하였던 파일 포맷이 아닌 아예 새롭게
qza/qzv를 만들어서 사용한다는... 음..
qiime2를 설치하지않은 사용자들을 위한 qza/qzv view사이트까지
(모하자는거지... 유료화로 바꿀건가)
그리고 CLI기반이 아닌 q2studio라는 GUI 인터페이스를 제공한다는
기쁜소식!! (인지 아닌지는 까봐야..)
GUI는 자동화하기 귀찮은데... 제길..
qiime1보다 report가 얼마나 세련되졌는 확인할 필요가 있고
그에 따라 유료화를 점쳐볼수도...
그러므로 나는 일단 소스 다운로드;;;;
설치만 해두려고 qiime2 설치하다가 환경 설정이 꼬였는지
쓰고있던 qiime 1이 작동안해서 멘붕이 왔다는건 비밀;;; 빼고는
아직까지는 호감인 녀석!
여튼 이번 qiime2 출시하면서 변화를 준게 완전히 qiime 플랫폼을 만든듯..
통상적으로 사용하였던 파일 포맷이 아닌 아예 새롭게
qza/qzv를 만들어서 사용한다는... 음..
qiime2를 설치하지않은 사용자들을 위한 qza/qzv view사이트까지
(모하자는거지... 유료화로 바꿀건가)
그리고 CLI기반이 아닌 q2studio라는 GUI 인터페이스를 제공한다는
기쁜소식!! (인지 아닌지는 까봐야..)
GUI는 자동화하기 귀찮은데... 제길..
qiime1보다 report가 얼마나 세련되졌는 확인할 필요가 있고
그에 따라 유료화를 점쳐볼수도...
그러므로 나는 일단 소스 다운로드;;;;
수요일, 11월 02, 2016
16S rRNA Variable regions
 |
| Variable regions of the 16S ribosomal RNA. |
화요일, 10월 25, 2016
PacBio SMRT Portal 설치
우선 서버사양은 Dell Workstation인데
CPU대비 메모리가 풍족해서 아직 메모리로 문제가 된적은 없는
Ubuntu 16.04.1 LTS 로 생정보하는데 필요한 프로그램이 설치되어 있고
관련 라이브러리가 설치된 조건하에서
SMRT Portal 설치를 시도하였고 전혀 Error없이 설치!! :)
SMRT Analysis Download 페이지에서 하단의 run 파일 두개 다운로드
무조건 2개 모두 다운로드!!
설치필수파일: smrtanalysis_2.3.0.140936.run
패치파일: smrtanalysis-patch_2.3.0.140936.p5.run
다운로드 후에
smrtanalysis 유저와 동일한 이름의 유저그룹도 생성한 후 설치 진행하시면되겠습니다.
$su -l smrtanalysis
$bash smrtanalysis_2.3.0.140936.run -p smrtanalysis-patch_2.3.0.140936.p4.run --rootdir
/path/to/smrtanalysis
설치에 필요한 프로그램들 번들로 설치하고 기존에 mysql 서버 사용중이라도 영향을 미치지않게 포트도 별도로 사용합니다. tomcat의 경우는 기존에 설치되어 있는 서비스가 있다면 영향을 미칠지 명확하지는 않습니다.(아마 포트를 변경해서 영향이 최소화되도록 진행될것 같습니다.)
설치 후 localhost가 아닌경우 방화벽 포트 열어주시면 웹페이지로 접속하여
멋있는 SMRT portal home 화면을 보실수 있으실겁니다. :)
ufw allow [설치할때 보였던 port들]
참고문헌 [Installation SMRT Portal]
피드 구독하기:
글 (Atom)