하는일이 이거다보니.. :)
여튼 오늘은 16S rRNA와 시퀀싱플랫폼에 대해서 잠시 이야기 하도록 하겠습니다.
최근 16S rRNA sequencing의 최강자 454가 서비스를 bye bye한 관계로
많은 연구자들이 MiSeq체제로 변환하고 있는데 (물론 Ion도 있고, PacBio도 있습니다.)
기존에 454를 사용했을 때와 다른 V region을 사용하고 다소 다른 결과들을 보이는 것들이
있을 것 입니다. 그래서 이리저리 검색하다가 걸린 논문 두 개를 가지고 잠시 얘기해보고자 합니다.
Nucl. Acids Res. (2010) 38 (22): e200.
PeerJ (2016) 4:e1869
논문을 찾아보기 시작한 이유는 454가 막을 내린 후 다른 시퀀싱 플랫폼에서는 왜 다른 region을 target하고 있고 왜 diversity에 차이를 보이는지... (내가 분석을 잘못했나.. ㅎㄷㄷㄷ)
우선 V region, 454는 V1-V2였는데 MiSeq은 V4 region을, Ion은 4개 region? 7개 region? 을 동시에target하고 있다는..
모 논문보면 아시겠지만
2010년 논문은 454 vs MiSeq을 비교했습니다. 두 플랫폼이 차이가 날까? 어떤것이 차이가 날까?
2010년도 논문을 한장 figure 요약하자면 이거죠

상단은 phylum abundance/ 하단은 genus abundance 그리고 좌측에 있는 V4는 이전 연구에서 사용되었던 기준이라고 생각되는 참고용 분포입니다. 실험 결과가 V4 region이 저 분포를 나타내면 실험이 잘됐다고 확인하는 용도로 사용됩니다.
사실 phylum은 크게 차이없죠, 차이가 있기도 쉽지않습니다.
관심사는 genus되겠습니다. 일루미나 데이터의 대부분은 unclassified입니다. 논문에서는 error때문에 이렇게 나왔으니 error좀 낮아지면 일루미나가 output이 많으니 sequence error 문제점을 개선된다면 미생물 분석에 적합한 킹왕짱 시퀀서가 될거라고 하는데 개인적인 생각으로는 sequence error(454가 일루미나가 한테 sequence error ㅋㅋ 좀 웃겼다능..
systematic error는 눈에 안뵈냐라고 한다면 눼눼, 하긴 이 논문이 2010년이란 것을 감안한다면 무리는 아니긴 합니다.)보다는 db선택이 unclassified문제는 보정할 수 있지 않을까 합니다.
그렇다면 이제 16s rRNA 입문한지 얼마안된 님께서 그렇게 느끼는 느낌적인 이유는
무엇인가?
바로 2016년 논문되겠습니다.
2016년 논문은 PacBio로 시퀀싱한 것을 db에 따라 분석 결과가 달라지는냐에 대한 내용으로
다음 한 장으로 요약 할 수 있겠습니다.
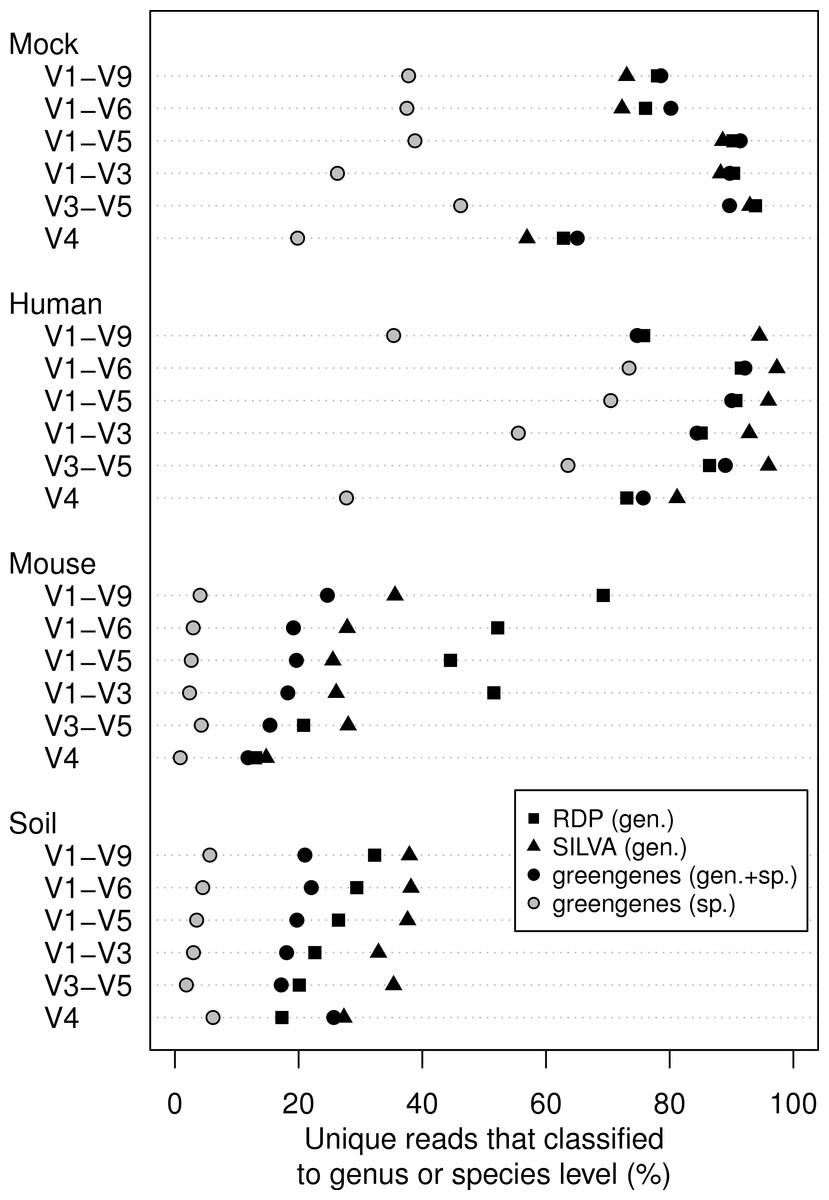
동일한 시료를 가지고 시퀀싱한 데이터를 16S rRNA db에 따라 classification되는 정도를 확인해본 그래프 입니다. 느낌 딹오시죠?
다만 나중에 뒤통수 맞았다는 느낌 안 받게 하나 말씀드리자면 RDP와 Silva의 경우 genus까지만 확인했고 gg는 genus와 genus+species 두가지로 확인한 것 입니다.
이제 PacBio의 Sequel에서 CCS로 생산된 16S rRNA 서열가지고 연구해도 나쁘지 않을 것 같다는..
아.. 이제 돈만 많으면 되는건가!!!