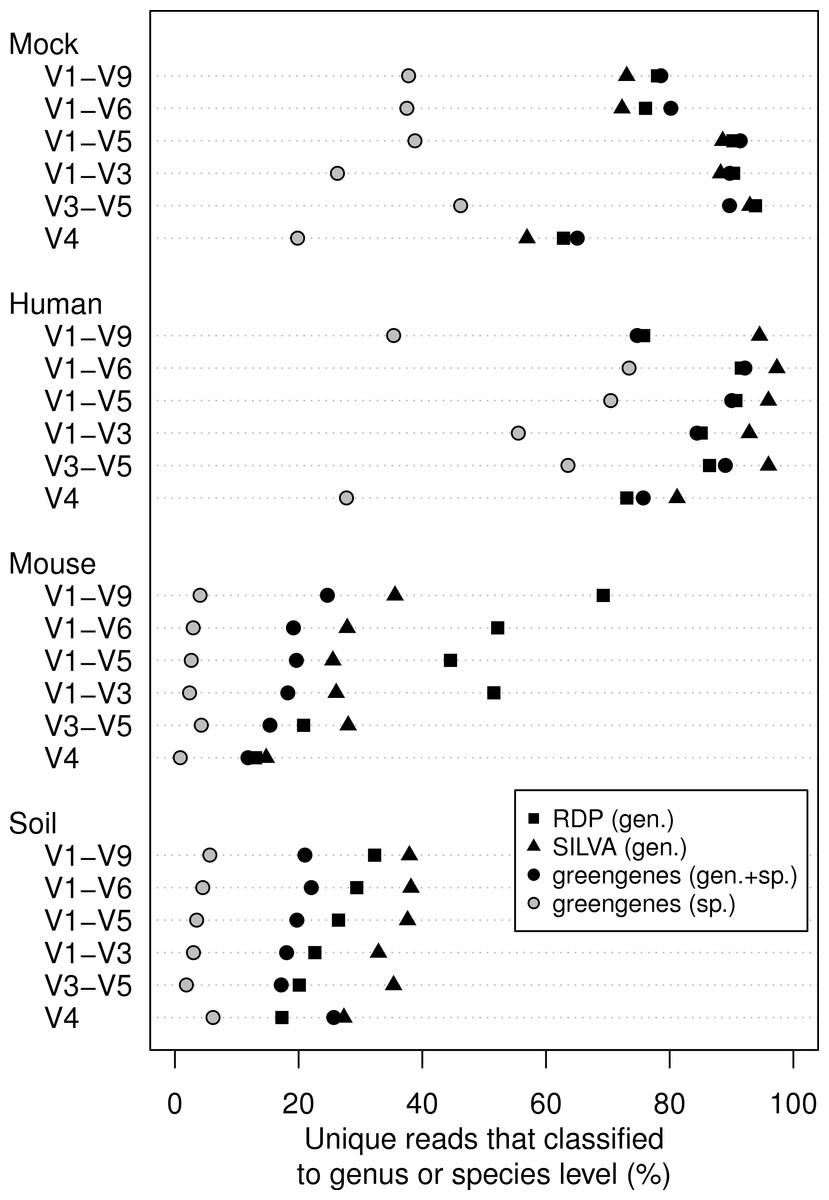
microbiome분석시 사용하는 reference database에 관련된 내용으로 SILVA, RDP, Greengenes, NCBI and OTT — how do these taxonomies compare? 2017년도에 나왔던 것으로 사실 지금은 greengene2의 등장으로 대부분 해소가 되었을 것이긴 하나 그래도 아직 완벽하게 gg2로 모든것이 해결된것은 아니라서...
gg2가 주로 사용되고 있긴하지만 그래도 다른 DB들을 아예 사용하지 않는 것은 아니라서 SILVA, RDP, greengene, NCBI, OTT에 대해서 한번 둘러보고, 각 DB별로 어떤 특장점들이 있는지 알아가는 기회가 되면 좋을것 같습니다.
DOI: 10.1186/s12864-017-3501-4
clova-x
이 연구는 microbiome sequencing 분석에서 사용되는 다양한 taxonomy의 호환성을 비교하고, 이를 통해 서로 다른 분류 체계 간의 분석 결과를 비교할 수 있는 방법을 제시합니다.1. **연구 배경**- microbiome sequencing 분석의 주요 단계 중 하나는 reads를 taxonomic units에 할당하는 것입니다.- SILVA, RDP, Greengenes, NCBI 등 네 가지 taxonomy가 주로 사용됩니다.2. **연구 방법**- taxonomy 간의 호환성을 평가하기 위해 mapping 알고리즘을 개발하였습니다.- 이 알고리즘을 통해 서로 다른 taxonomy 간의 mapping을 수행하고, 분석 결과를 비교할 수 있습니다.3. **결과 및 결론**- SILVA, RDP, Greengenes는 NCBI와 OTT에 잘 매핑되지만, 반대 방향으로는 문제가 발생합니다.- 가장 큰 두 taxonomy인 NCBI와 OTT는 작은 taxonomy들에 비해 매핑이 어렵습니다.4. **Taxonomy 설명**- SILVA는 bacteria, archaea, eukarya에 대한 정보를 포함하며, 16S/18S rRNA를 기반으로 합니다.- RDP는 16S rRNA sequences를 기반으로 하며, bacteria, archaea, fungi에 대한 정보를 포함합니다.- Greengenes는 bacteria와 archaea에 초점을 맞추고, 자동화된 tree construction을 사용합니다.- NCBI는 모든 유기체의 이름을 포함하며, 매일 업데이트됩니다.- OTT는 가능한 한 많은 taxa를 포괄하는 종합적인 tree를 제공하며, phylogenetic trees를 기반으로 합니다.
gemini
## 제목: 미생물 분류의 혼란을 해결하다: SILVA, RDP, Greengenes, NCBI 그리고 OTT는 어떻게 다를까요?**연구 배경: 미생물 분류, 왜 중요하고 무엇이 문제일까요?**[cite_start]최근 '마이크로바이옴(Microbiome)'이라고 불리는 우리 몸과 환경 속 미생물에 대한 연구가 활발합니다[cite: 596]. [cite_start]미생물의 DNA 염기 서열을 분석하는 것은 이들 미생물의 구성을 파악하는 핵심 단계입니다[cite: 588, 597]. [cite_start]이러한 분석에서 미생물을 정확하게 분류하고 이름을 붙이는 기준, 즉 **'참조 분류 체계(Reference Taxonomy)'**의 선택이 매우 중요합니다[cite: 598, 601].[cite_start]문제는 미생물 분류를 위해 널리 사용되는 분류 체계, 예를 들어 **SILVA, RDP, Greengenes, NCBI**가 서로 다르고 일관성이 부족하다는 점입니다[cite: 589, 601]. [cite_start]연구자들은 이 중 하나를 선택해 분석을 진행하지만, 각 분류 체계가 얼마나 비슷한지, 그리고 다른 분류 체계를 사용한 연구 결과를 어떻게 비교해야 할지 명확하지 않았습니다[cite: 590, 604].**연구 목적: 미생물 분류 체계들을 비교하고 호환성을 확보하자**[cite_start]이 논문은 널리 사용되는 네 가지 미생물 분류 체계(SILVA, RDP, Greengenes, NCBI)와 최근 발표된 **Open Tree of Life Taxonomy (OTT)**를 비교하여[cite: 592, 602, 607]:1. [cite_start]이들 분류 체계가 얼마나 유사한지를 파악하고 [cite: 602]2. [cite_start]하나의 분류 체계를 사용한 결과를 다른 분류 체계로 쉽게 변환하여 비교할 수 있는지 확인하는 것 [cite: 604, 606]을 목표로 합니다.**연구 방법: 분류 체계 간의 '매핑(Mapping)'과 유사성 점수**연구진은 분류 체계들을 비교하기 위해 두 가지 주요 접근 방식을 사용했습니다.1. **공유 분류 단위 비교 (Shared Taxonomic Units):*** [cite_start]Phylum(문)부터 Genus(속) 수준까지, 각 분류 체계가 이름이 동일한 분류 단위를 얼마나 공유하고 있는지 세어 비교했습니다[cite: 710, 714].* [cite_start]특히, 네 가지 분류 체계의 합집합('ALL')과 OTT를 비교했습니다[cite: 715].2. **매핑 절차 (Mapping Procedure) 기반 비교:*** [cite_start]하나의 분류 체계(출발지 A)의 노드(분류 단위)를 다른 분류 체계(도착지 B)의 노드로 연결하는 '매핑' 방법을 정의하고 소프트웨어(**CrossClassify**)로 구현했습니다[cite: 591, 717, 718, 847].* [cite_start]**'엄격한 매핑(Strict mapping)'**과 **'느슨한 매핑(Loose mapping)'**을 사용하여 A의 노드가 B에서 동일한 이름과 등급(Rank)을 가진 노드를 찾지 못했을 때의 처리 방식을 다르게 했습니다[cite: 720, 726, 834].* 매핑이 완벽하지 않을 때, 두 분류 체계의 노드 등급 차이를 이용해 **'비유사성 점수(Dissimilarity Score, $Q(A,B)$)'**를 계산했습니다. [cite_start]이 점수는 0(완벽한 매핑)부터 1(모든 노드가 뿌리 노드에 매핑) 사이의 값을 가집니다[cite: 814, 816, 817, 842].**연구 결과: 큰 분류 체계가 작은 분류 체계를 잘 포용한다**1. **공유 분류 단위:*** [cite_start]네 가지 분류 체계(SILVA, RDP, Greengenes, NCBI)는 Genus(속) 수준에서 무려 89%의 분류 단위가 다른 분류 체계와 공유되지 않는 등, 분류 단위의 차이가 컸습니다[cite: 928].* [cite_start]SILVA는 NCBI와 가장 많은 분류 단위를 공유했습니다 (Phylum, Class, Order에서 60%, Family, Genus에서 10%)[cite: 929].* [cite_start]다른 세 가지 분류 체계(SILVA, RDP, Greengenes)의 교집합이 NCBI를 제외하고는 매우 작았는데, 이는 이들 세 분류 체계가 대부분 NCBI 분류 체계에 포함됨을 시사합니다[cite: 930].2. **매핑 비교:*** [cite_start]**SILVA, RDP, Greengenes**와 같은 작은 분류 체계는 **NCBI와 OTT**와 같은 큰 분류 체계로 **잘 매핑**되었습니다 (낮은 비유사성 점수)[cite: 593, 608, 937, 938].* [cite_start]특히 NCBI와 OTT는 평균 비유사성 점수가 각각 0.08과 0.06으로 매우 낮았습니다[cite: 938].* [cite_start]반면, **NCBI와 OTT**와 같은 큰 분류 체계는 작은 분류 체계로 매핑할 때 **문제가 많았습니다** (평균 비유사성 점수 약 0.68로 훨씬 높음)[cite: 593, 608, 939].* [cite_start]가장 큰 **OTT**는 **NCBI**에 매핑할 때 (비유사성 0.12)보다 **NCBI**가 **OTT**에 매핑할 때 (비유사성 0.02) 훨씬 더 유사성이 높았습니다[cite: 940].**고찰 및 의의와 시사점: NCBI를 공통의 언어로 사용하자*** [cite_start]**크기 효과:** 단순한 공유 분류 단위 비교는 분류 체계의 크기에 크게 좌우되어 구조적 호환성에 대한 정보를 거의 제공하지 못했습니다[cite: 1004, 1006].* [cite_start]**구조적 호환성:** 매핑 분석은 **SILVA, RDP, Greengenes**가 **NCBI와 OTT**에 비교적 잘 포용된다는 것을 보여주어, 이들 작은 분류 체계의 연구 결과를 더 큰 분류 체계로 변환하는 것이 가능하다는 것을 시사합니다[cite: 593, 608].* **분류 체계 선택의 중요성:*** [cite_start]OTT는 다섯 가지 분류 체계 중 가장 크고 다른 분류 체계들이 잘 매핑되지만, 현재는 서열 데이터베이스가 없어 미생물 유전체학(Metagenomics) 연구에 직접 사용하기에는 한계가 있습니다[cite: 1048, 1049].* [cite_start]따라서, 이 연구는 **다른 분류 체계를 사용하여 수행된 분석들을 비교할 때 공통의 프레임워크로 NCBI 분류 체계를 사용할 것을 권장**합니다[cite: 1050].* [cite_start]16S 서열 분석만 수행하는 연구에서도 널리 사용되는 SILVA 대신 NCBI 분류 체계를 사용하거나, 16S 및 샷건 시퀀싱을 모두 사용하는 연구에서는 NCBI를 고려해야 합니다[cite: 1051].**이 연구가 중요한 이유**[cite_start]이 연구는 서로 다른 미생물 분류 체계 간의 복잡하고 구조적인 유사성을 정량적으로 비교할 수 있는 **새로운 방법과 소프트웨어(CrossClassify)**를 제시했습니다[cite: 591, 605, 606, 847]. [cite_start]미생물 분류의 일관성 부족은 마이크로바이옴 연구의 결과를 통합하고 비교하는 데 큰 걸림돌이었습니다[cite: 590, 604]. [cite_start]이 연구의 결과와 도구는 연구자들이 서로 다른 분류 체계 기반의 **마이크로바이옴 연구 결과를 공통의 분류 체계로 통일하여 비교할 수 있도록** 지원함으로써, 향후 연구의 **호환성과 재현성을 크게 향상**시키는 데 기여할 것입니다[cite: 606].
chatGPT
다음은 업로드하신 논문 **「SILVA, RDP, Greengenes, NCBI and OTT — How do these taxonomies compare?」(BMC Genomics, 2017)**의 내용을 일반 독자가 이해하기 쉽게 정리한 블로그용 요약입니다.(A4 두 장 분량, 연구 배경 → 목적 → 방법 → 결과 → 고찰 → 의의 순서)---## 미생물 분류의 ‘지도’를 비교하다 — 다섯 가지 미생물 분류체계의 차이점### 🧬 연구 배경우리 몸이나 환경 속의 미생물을 분석할 때, 연구자들은 ‘누가 살고 있는가’를 알아내기 위해 **DNA를 직접 시퀀싱(염기서열 분석)** 합니다.하지만 여기서 큰 문제가 하나 있습니다.분석한 DNA 조각들을 ‘어떤 생물 분류체계(taxonomy)’를 기준으로 분류하느냐에 따라 **결과가 달라질 수 있다는 것**입니다.현재 미생물 연구자들이 가장 널리 쓰는 분류체계는 다음 네 가지입니다.* **SILVA*** **RDP (Ribosomal Database Project)*** **Greengenes*** **NCBI Taxonomy**최근에는 여기에 **Open Tree of Life Taxonomy (OTT)** 라는 방대한 계통수가 새로 등장했습니다.이들 분류체계는 모두 ‘미생물의 계통학적 이름표’를 제공하지만, 구성 방식과 깊이, 세분화 수준이 제각각입니다.그래서 서로 다른 연구에서 결과를 비교하거나 통합하기가 매우 어렵습니다.---### 🎯 연구 목적이 논문은 **이 다섯 가지 분류체계가 얼마나 유사하거나 다른지**를 분석했습니다.즉, “같은 미생물이라도 다른 분류체계에서는 이름과 위치가 다르게 나타나는 이유는 무엇일까?”를 탐구한 것입니다.이를 통해 연구자들이 **서로 다른 분류체계를 쓴 연구 결과를 공정하게 비교할 수 있는 방법**을 제시하고자 했습니다.---### 🧩 연구 방법연구팀은 **“CrossClassify”라는 소프트웨어**를 만들어,한 분류체계의 생물 이름을 다른 분류체계로 **‘매핑(mapping)’** 할 수 있도록 했습니다.매핑에는 세 가지 방식이 쓰였습니다.1. **엄격한 매핑(strict mapping)** – 이름과 계급(문, 강, 목 등)이 완벽히 일치할 때만 연결.2. **느슨한 매핑(loose mapping)** – 일부 단계가 달라도 가능한 한 가까운 수준으로 연결.3. **경로 비교(path comparison)** – 동일한 생물로 이어지는 계통 경로가 얼마나 비슷한지 비교.이 과정을 통해 각 분류체계의 구조적 유사도(또는 차이)를 수치화했습니다.---### 🔬 연구 결과1. **SILVA, RDP, Greengenes는 NCBI와 OTT에 잘 매핑된다.**→ 이 네 체계는 대부분 NCBI에 포함되거나 유사한 구조를 가집니다.2. **하지만 NCBI나 OTT를 반대로 작은 체계(SILVA·RDP·Greengenes)에 매핑하면 오류가 많다.**→ 큰 지도(OTT)를 작은 지도 위에 억지로 맞추려는 셈이죠.3. **Greengenes는 가장 단순하고 오래되어 불일치가 많다.**→ 2013년 이후 업데이트되지 않아 현재는 최신 연구에 부적합합니다.4. **NCBI와 OTT는 매우 유사하다.**→ OTT가 더 크지만, 구조적으로 거의 일치(불일치율 < 2%)했습니다.5. **결론:** SILVA·RDP·Greengenes보다 **NCBI가 중심 기준으로 사용하기 가장 적합**합니다.---### 💡 고찰 및 시사점미생물 연구에서 분류체계는 ‘지도’와 같습니다.다섯 가지 지도 중 어떤 것을 쓰느냐에 따라 **같은 데이터를 다르게 해석할 수** 있습니다.예를 들어, 한 연구에서 “어떤 세균이 많다”고 한 결과가 다른 분류체계에서는 이름이 달라지거나 아예 존재하지 않을 수도 있습니다.이 논문은 “지도 간의 차이”를 정량화하고, 이를 자동으로 변환할 수 있는 도구를 제시했습니다.이를 통해 연구자들은 **다른 분류체계를 쓴 연구들을 비교·통합·검증**할 수 있게 되었습니다.또한, 미생물 데이터베이스 업데이트와 표준화의 중요성을 강조했습니다.---### 🌍 연구의 의의와 중요성이 연구는 단순히 데이터베이스를 비교한 것이 아니라,**전 세계 미생물 연구의 ‘공통 언어’를 만드는 기초 작업**이라 할 수 있습니다.* 앞으로 수많은 미생물 유전체 연구가 쏟아질 텐데,이들을 정확히 비교하려면 신뢰할 수 있는 기준이 필요합니다.* 이 연구가 제시한 매핑 방법은 서로 다른 연구 결과를 하나의 체계로 통합할 수 있는 **‘번역기’ 역할**을 합니다.* 궁극적으로는 **인체 마이크로바이옴, 환경 미생물 연구, 신약 개발** 등 다양한 분야에서연구 간의 일관성을 높이고 데이터 활용도를 극대화할 수 있습니다.---요약하자면,이 논문은 “미생물 분류체계 간의 경계선을 비교해, 서로 다른 지도들을 하나로 읽을 수 있게 만든 연구”입니다.그 결과 **NCBI Taxonomy를 중심으로 통합 연구를 진행하는 것이 가장 합리적**이라는 결론을 제시했습니다.
chatGPT (스토리텔링)
아래는 논문 **〈SILVA, RDP, Greengenes, NCBI and OTT — How do these taxonomies compare?〉**를 기반으로 한스토리텔링형 블로그 글입니다.전문적 내용은 유지하되, 일반 독자가 흥미롭게 읽을 수 있도록 서사와 비유를 곁들였습니다.---# 🧬 다섯 개의 미생물 지도, 누가 진짜 표준인가?“같은 장소를 가리키는데, 지도마다 위치가 다르다면?”이런 상황이 실제로 **미생물 연구 세계**에서 벌어지고 있습니다.과학자들은 인체, 토양, 바다 속에 사는 미생물들의 DNA를 분석해‘누가 살고 있는지’를 밝히려 하지만,문제는 **그 결과를 어디에 맞춰 해석하느냐**에 따라 달라진다는 것입니다.그 이유는 바로 —세상에는 서로 다른 **다섯 개의 미생물 분류 지도**,즉 **SILVA, RDP, Greengenes, NCBI, OTT**가 존재하기 때문이죠.---## 🗺️ 서로 다른 지도, 혼란스러운 미생물 세계미생물 연구에서 가장 중요한 단계 중 하나는DNA 조각(시퀀스)을 읽어들인 뒤,그 조각이 **어떤 생물**에 속하는지를 분류하는 일입니다.이때 과학자들은 특정 **분류체계(taxonomy)**를 기준으로“이건 세균 ○○속(genus)에 속한다” 식으로 이름을 붙입니다.그런데 분류체계가 여러 개인 탓에,같은 미생물도 어떤 연구에서는 ‘A속’, 다른 연구에서는 ‘B속’으로 불리기도 합니다.예를 들어, 우리가 ‘서울’이라 부르는 도시를어떤 지도에서는 ‘한성’, 어떤 지도에서는 ‘Seoul City’로 적어두는 셈이죠.이렇게 이름이 제각각이면 연구자들끼리 결과를 비교하기가 매우 어렵습니다.그래서 튀빙겐 대학의 **모니카 발보추테(Monika Balvočiūtė)**와 **다니엘 휴손(Daniel Huson)**은이 문제를 정면으로 다루었습니다.그들의 목표는 명확했습니다.> “다섯 개의 미생물 지도가 얼마나 닮았는가,> 그리고 그 차이를 어떻게 하나의 기준으로 묶을 수 있을까?”---## 🔍 연구진의 도전: 서로 다른 분류체계를 이어 붙이다연구팀은 각 분류체계를 서로 ‘매핑(mapping)’하는새로운 방법과 프로그램을 만들었습니다.이 프로그램의 이름은 **CrossClassify** —말 그대로 “분류체계를 교차시켜 비교한다”는 뜻이죠.그들은 세 가지 방식으로 각 분류체계를 연결했습니다.1. **엄격한 매핑(strict)*** 이름과 단계(문, 강, 목 등)가 정확히 일치할 때만 연결.* 가장 보수적인 방식입니다.2. **느슨한 매핑(loose)*** 일부 단계가 달라도 비슷한 위치라면 연결.* 실제 연구 상황에 가까운 비교입니다.3. **경로 비교(path)*** 같은 생물로 이어지는 ‘가지 구조(계통)’가 얼마나 유사한지 평가.이 과정을 통해 연구진은“각 지도 간의 거리”를 수치로 계산할 수 있게 되었습니다.---## 📊 결과: 가장 넓고 안정적인 지도는 ‘NCBI’분석 결과는 흥미로웠습니다.* **SILVA, RDP, Greengenes**는 대부분 **NCBI**와 잘 맞았습니다.→ 즉, NCBI는 다른 지도들과 겹치는 영역이 많았습니다.* 반대로 **NCBI나 OTT(Open Tree of Life Taxonomy)** 같은 큰 체계를작은 체계로 매핑하려 하면 충돌이 많았습니다.→ 큰 지도를 작은 지도 위에 억지로 덮어씌운 셈이죠.* **Greengenes**는 특히 불일치가 컸습니다.→ 2013년 이후 업데이트가 중단되어, 이미 ‘낡은 지도’가 되어버렸습니다.* **OTT와 NCBI는 거의 완벽히 일치(불일치율 2% 미만)**했습니다.→ OTT는 더 방대한 버전의 NCBI라 볼 수도 있죠.연구팀은 결론을 이렇게 요약했습니다.> “미생물 연구에서 서로 다른 결과를 비교하고 통합하려면> **NCBI 분류체계를 기준**으로 삼는 것이 가장 바람직하다.”---## 🧭 왜 이 연구가 중요한가?이 연구는 단순히 데이터베이스를 비교한 게 아닙니다.미생물학의 근본적인 문제,즉 “**서로 다른 연구가 같은 언어로 소통할 수 있는가**”를 다룬 것입니다.예를 들어, 한 연구팀이 사람 장내 세균을 NCBI 기준으로 분석하고,다른 팀이 SILVA 기준으로 분석했을 때같은 세균이 서로 다른 이름으로 나올 수 있습니다.그럼 두 결과를 비교하거나 통합하는 게 불가능하죠.하지만 이 논문에서 제시한 방법을 쓰면,서로 다른 분류체계 간의 대응 관계를 자동으로 계산해하나의 공통 기준으로 바꿔줄 수 있습니다.일종의 **‘미생물 세계의 번역기’**인 셈입니다.---## 🌍 앞으로의 의미이 연구는 미생물 연구의 ‘표준화 시대’를 여는 첫걸음이라 할 수 있습니다.NCBI를 기준으로 데이터를 통합하면전 세계의 연구자들이 같은 기준으로 미생물 생태계를 분석할 수 있고,이는 곧 **질병 연구, 신약 개발, 환경 생태 연구**의 정밀도를 높여줍니다.‘다섯 개의 미생물 지도’는 이제 하나의 공통 언어를 찾았습니다.그리고 그 중심에는,**가장 크고 정교한 지도 — NCBI Taxonomy**가 있습니다.---🧠 **정리하자면:**이 연구는 “어떤 미생물 지도(분류체계)를 써야 할까?”라는 오랜 의문에명확한 해답을 준 논문입니다.다양한 데이터와 도구가 넘쳐나는 지금,정확한 기준을 세우는 일은 모든 과학의 출발점이죠.> 미생물학자들에게 이 연구는,> ‘지도 제작의 혼돈’을 끝내고 ‘공통의 나침반’을 제공한 역사적인 작업이었습니다.
grok3
### 미생물 분류 데이터베이스 비교: SILVA, RDP, Greengenes, NCBI, OTT#### 연구 배경미생물 군집 분석(마이크로바이옴 시퀀싱)은 세균이나 곰팡이 같은 미생물을 실험실에서 키우지 않고 DNA를 직접 분석하는 기술입니다. 이 과정에서 DNA 시퀀싱 데이터를 '분류학 단위'(예: 균종, 속, 과)로 배정하는 게 핵심인데, 보통 SILVA, RDP, Greengenes, NCBI라는 네 가지 데이터베이스를 사용합니다. 하지만 이 데이터베이스들이 서로 얼마나 비슷한지, 다른 걸 썼을 때 결과를 어떻게 비교할지 불분명합니다. 이 논문은 이런 문제를 해결하려고 시작됐어요. 추가로 최근 나온 OTT(생명나무 열린 분류학)도 포함해 비교합니다.#### 연구 목적네 가지 주요 분류학(SILVA, RDP, Greengenes, NCBI)과 OTT를 비교하고, 하나의 분류학을 다른 것으로 '매핑'(대응시키기)하는 방법을 개발하는 게 목표입니다. 이를 통해 서로 다른 데이터베이스를 쓴 분석 결과를 공통적으로 비교할 수 있게 하려 해요. 궁극적으로 미생물 연구에서 분류학 선택의 중요성을 밝히고, 호환성을 높이는 도구를 제공합니다.#### 연구 방법먼저 각 분류학의 크기와 구조를 분석했습니다. SILVA는 12,117개 노드(분류 단위), RDP는 6,128개, Greengenes는 3,093개, NCBI는 1,522,150개, OTT는 2,627,066개로 크기가 다양해요. 주요 계급(도메인, 문, 강, 목, 과, 속, 종)만 고려했습니다.비교 방법:- **공유 단위 비교**: 각 계급에서 이름이 같은 단위를 세서 겹침 정도를 봅니다. (NCBI 동의어 사전으로 이름 통일)- **매핑 절차**: 한 분류학의 노드를 다른 분류학에 대응시킵니다.- 엄격 매핑: 위에서 아래로 완벽 일치할 때만 매핑, 아니면 부모 노드로.- 느슨 매핑: 아래에서 위로, 일부 불일치 허용.- 경로 비교: 공통 노드의 뿌리부터 경로만 비교.- 불일치 점수(Q): 매핑된 노드의 계급 차이를 계산해 전체 유사도 측정 (0: 완벽, 1: 완전 불일치).이 방법을 Java 프로그램 'CrossClassify'로 구현해 BIOM 형식 파일 처리 가능하게 했습니다.#### 연구 결과- 공유 단위: 네 분류학 중 NCBI가 SILVA와 가장 많이 겹칩니다(문/강/목 계급 60%). 전체 네 개의 합집합(ALL)은 OTT와 목/과 계급에서 59%/46% 공유하지만, 속/종은 OTT가 더 다양합니다.- 매핑 결과: SILVA, RDP, Greengenes는 NCBI/OTT로 잘 매핑(불일치 0.06~0.21)되지만, 반대(큰 걸 작은 걸로)는 어려움(0.58~0.77). NCBI와 OTT는 서로 잘 맞아요(0.02~0.12). 경로 비교는 불일치가 적지만(중앙값 0.02), 공유 구조에도 약간 차이 있음.- 시각화: 히트맵과 병렬 세트로 불일치 패턴 보임. 예: Greengenes에서 Persicus 종 경로 매핑 예시.#### 고찰크기 차이(OTT/NCBI가 훨씬 큼)가 매핑 어려움의 주원인입니다. 엄격 매핑은 코어 구조 불일치를, 느슨 매핑은 전체 호환성을 보여줘요. OTT는 크지만 시퀀스 데이터 없어 메타게놈 연구에 제한적. SILVA/RDP/Greengenes는 16S rRNA 기반으로 비슷하지만, Greengenes는 업데이트 안 돼 덜 다양. NCBI는 16S와 샷건 시퀀싱 모두에 적합해 공통 프레임워크로 추천합니다. 추가 응용: 다른 빈닝 방법(예: LCA vs. wLCA) 비교에도 유용.#### 의의와 시사점이 연구는 분류학 간 매핑 도구를 제공해, 서로 다른 데이터베이스를 쓴 미생물 연구 결과를 비교 가능하게 합니다. 예를 들어, SILVA 기반 분석을 NCBI로 변환할 수 있어요. 미생물 분류의 불일치(8% 알려짐)를 고려하면, 연구자들이 더 일관된 결과를 낼 수 있게 돕습니다. 시사점으로는 OTT처럼 포괄적 분류학이 미래지만, NCBI처럼 실용적인 걸 우선 쓰라는 조언입니다.이 연구가 중요한 이유는 미생물 군집 분석이 건강, 환경, 농업 등에 핵심인데, 분류학 차이로 결과가 왜곡될 수 있기 때문입니다. 호환성을 높여 연구 신뢰성을 강화하고, 더 정확한 미생물 이해를 돕습니다. (약 850자, A4 1.5장 분량)
NotebookLM
## 논문 요약: SILVA, RDP, Greengenes, NCBI, OTT — 이 분류 체계들은 어떻게 비교될까요?**대상 독자:** 미생물 유전체 분석에 관심 있는 일반 성인 독자---### 연구 배경: 미생물 분석의 기준점을 찾아서최근 건강과 환경 분야에서 **마이크로바이옴**(특정 환경에 서식하는 미생물 군집) 연구가 활발합니다. 이 연구는 미생물을 실험실에서 배양하지 않고 DNA 염기서열만을 분석하여 미생물의 종류와 구성을 파악합니다.이 분석 과정에서 가장 중요한 단계 중 하나는 수많은 염기서열 조각들을 해당하는 **분류 단위(taxonomic units)**로 할당하는 것입니다. 이때 미생물을 분류하는 기준이 되는 **분류 체계(taxonomy)**가 필요합니다. 현재 미생물 염기서열 분석, 특히 16S rRNA 유전자 분석에서는 주로 네 가지 분류 체계, 즉 **SILVA, RDP, Greengenes, NCBI** 중 하나를 사용하고 있습니다.문제는 이 네 가지 분류 체계가 서로 얼마나 유사한지, 그리고 서로 다른 분류 체계를 사용해 도출된 분석 결과를 어떻게 신뢰성 있게 비교할 수 있는지 명확하지 않다는 점입니다. 이 연구는 이러한 불확실성을 해소하고자 시작되었습니다.### 연구 목적: 분류 체계 간 호환성 확인이 논문의 주된 목적은 널리 사용되는 네 가지 미생물 분류 체계(SILVA, RDP, Greengenes, NCBI) 간의 유사성을 정량적으로 확인하는 것입니다. 나아가, 한 분류 체계를 사용하여 얻은 분석 결과가 다른 분류 체계로 **쉽게 변환되거나 비교될 수 있는지** 알아보고자 했습니다. 이 연구에서는 이 네 가지 분류 체계와 함께 최근 발표된 방대한 규모의 **OTT (Open Tree of life Taxonomy)**도 함께 비교 대상으로 포함했습니다.### 연구 방법: 분류 체계 매핑 기법 개발연구진은 다섯 가지 분류 체계를 비교하기 위해 **한 분류 체계의 분류 단위를 다른 분류 체계로 대응(mapping)시키는 방법과 소프트웨어(CrossClassify)**를 개발했습니다.다섯 가지 분류 체계는 규모와 분류 수준(해상도)에서 큰 차이를 보입니다. 예를 들어, NCBI와 OTT는 종(Species) 이하 수준까지 분류하는 반면, SILVA와 RDP는 속(Genus) 수준까지만 분류합니다.연구진은 분류 체계의 계층적 구조를 기반으로 세 가지 매핑 절차를 정의했습니다:1. **엄격한 매핑(Strict mapping):** 완벽하게 일치하는 분류 단위를 찾지 못하면 상위 조상으로 매핑합니다.2. **느슨한 매핑(Loose mapping):** 부분적으로 일치하는 경우에도 최대한 가까운 순위로 매핑하여 전반적인 호환성을 파악합니다.3. **경로 비교(Path comparison):** 이름과 순위가 동일한 분류 단위까지 이르는 경로(계층 구조)의 차이만을 비교합니다.이러한 매핑 결과를 기반으로, 두 분류 체계 간의 순위 배치 차이를 계산하여 **비유사도(dissimilarity)** 점수(0~1 사이 값)를 도출했습니다. 이 점수가 낮을수록 두 분류 체계가 호환된다는 의미입니다.### 연구 결과: NCBI와 OTT로의 일방향 매핑1. **분류 단위의 중복성:** 단순히 분류 단위의 이름만 비교했을 때, 네 가지 분류 체계(SILVA, RDP, Greengenes, NCBI)는 서로 공유하지 않는 고유한 분류 단위(예: 속(genus)의 89%)를 매우 많이 포함하고 있었습니다.2. **NCBI의 포괄성:** 흥미롭게도, SILVA, RDP, Greengenes 세 분류 체계의 교집합은 매우 작았는데, 이는 이 세 분류 체계의 분류 단위들이 **대부분 NCBI 분류 체계 안에 포함되어 있음**을 시사합니다.3. **매핑의 호환성:** 엄격한 매핑 결과, 대부분의 분류 체계 쌍에서 호환성이 낮았습니다 (비유사도 중앙값 0.5). 이는 분류 체계의 핵심 구조에 상당한 불일치가 있음을 보여줍니다.4. **일방향 매핑 성공:*** **SILVA, RDP, Greengenes**는 규모가 더 큰 **NCBI**나 **OTT**로 **잘 매핑되었습니다** (비유사도 점수가 낮게 나옴).* 그러나 그 반대 방향, 즉 NCBI나 OTT를 더 작은 분류 체계(SILVA, RDP, Greengenes)로 매핑하는 것은 **문제가 있었습니다** (비유사도 점수가 높게 나옴).5. **NCBI와 OTT의 유사성:** 두 가장 큰 분류 체계인 NCBI와 OTT는 서로 매우 유사하게 매핑되었습니다. 특히 NCBI를 OTT로 매핑했을 때 비유사도는 0.02로 매우 낮았습니다. 이 두 분류 체계는 경로 비교에서도 매우 흡사했습니다 (< 0.01). 다만, OTT가 NCBI보다 노드 수가 거의 두 배 많습니다.### 고찰 및 논의: 구조적 비교의 필요성연구진은 단순히 이름으로 분류 단위를 비교하는 것은 가장 작은 분류 체계의 크기에 좌우되기 때문에, **분류 체계의 구조적(위상적) 호환성**에 대해서는 거의 알려주지 못한다고 지적했습니다. 따라서, 이 연구에서 개발한 매핑 방법을 통해 분류 체계 간의 구조적 불일치를 정량적으로 파악하는 것이 중요합니다.엄격한 매핑과 느슨한 매핑 결과의 큰 차이는 분류 체계 간의 고위 순위(예: 문, 강)에서의 불일치가 흔하게 나타나지만, 전체적인 호환성은 느슨한 매핑을 통해 더 잘 파악할 수 있음을 보여줍니다.### 연구의 의의와 시사점: 공통 프레임워크의 제안이 연구는 서로 다른 미생물 분류 체계를 사용하여 얻은 분석 결과를 **통합적으로 비교하고 해석**할 수 있는 구체적인 방법론과 소프트웨어(CrossClassify)를 처음으로 제공했습니다.OTT는 다섯 가지 분류 체계 중 가장 크고 다른 모든 분류 체계들이 OTT로 잘 매핑된다는 점에서 이상적인 참조 분류 체계가 될 수 있습니다. 그러나 현재 OTT는 시퀀스 데이터베이스가 연결되어 있지 않아 메타게놈학 분석에서의 실용성이 제한됩니다.**따라서 연구진은 서로 다른 분류 체계를 사용하는 분석 결과를 비교할 때 NCBI 분류 체계를 공통 프레임워크로 사용할 것을 권고합니다.** 특히 16S 염기서열 분석과 전체 염기서열(샷건) 분석을 모두 사용하는 연구에서는 NCBI 분류 체계를 사용하는 것이 유리하다고 제안했습니다.---### 이 연구가 중요한 이유마이크로바이옴 연구가 의학, 생태학 등 다양한 분야에서 중요해지면서, 연구 결과의 **일관성과 재현성**이 핵심 과제가 되었습니다. 이 연구는 연구자들이 각자의 선호에 따라 사용하는 다양한 미생물 분류 체계(SILVA, RDP, Greengenes 등)가 서로 얼마나 다르고, 어떤 구조적 불일치를 가지고 있는지 **객관적인 수치와 수학적 모델**로 규명했습니다.이를 통해 연구자들은 서로 다른 분류 체계 기반의 분석 결과를 **표준화된 프레임워크(NCBI)로 통합**하고 비교할 수 있게 되었으며, 이는 마이크로바이옴 데이터 분석의 신뢰성과 상호 운용성을 획기적으로 향상시키는 기반이 됩니다. 또한, 이 매핑 기술은 미생물 분류 체계 자체를 비교하는 것 외에도, 염기서열 데이터를 분류 단위로 할당하는 **다양한 소프트웨어 방법론의 성능을 평가**하는 데에도 활용될 수 있어, 향후 분석 도구 개발에도 중요한 시사점을 제공합니다.
Perplexity.ai
이 논문 **“SILVA, RDP, Greengenes, NCBI, 그리고 OTT — 미생물 분류체계는 얼마나 다른가?”**(Balvočiūtė & Huson, 2017)은 미생물 유전체 연구에서 널리 사용하는 다섯 가지 분류체계의 차이를 체계적으로 비교한 연구이다.다음은 일반 독자도 쉽게 이해할 수 있도록, 연구의 핵심 내용을 A4용지 두 장 이내 분량으로 정리한 내용이다.***## 연구 배경최근 미생물 연구에서는 ‘미생물군집(마이크로바이옴)’ 분석이 중요하다. 예를 들어 우리의 장이나 토양 속에 존재하는 다양한 미생물의 DNA를 분석해 어떤 미생물이 얼마나 사는지를 알아내는 것이다. 이런 분석에서 가장 첫 번째 단계는 DNA 조각(읽힘, read)을 **어떤 생물 종에 속하는지** 분류하는 일인데, 이를 위해 미리 정리된 **참조 분류체계(taxonomy)** 가 필요하다.하지만 연구자마다 사용하는 분류체계가 다르다. 대표적인 다섯 가지는 **SILVA, RDP, Greengenes, NCBI, OTT(Open Tree of Life Taxonomy)** 이다. 문제는 이 다섯 시스템이 서로 이름과 구조가 조금씩 달라, 같은 데이터라도 분류체계에 따라 결과가 달라질 수 있다는 점이다. 이 논문은 “이들 분류체계가 얼마나 비슷하거나 다른가?”를 정량적으로 조사했다.***## 연구 목적이 연구의 목표는 다음 두 가지였다.1. **SILVA, RDP, Greengenes, NCBI, OTT** 다섯 가지 분류체계가 서로 얼마나 일치하는지 객관적으로 비교하기.2. 서로 다른 분류체계를 사용하는 연구 결과를 **비교 가능하게 만드는 방법과 소프트웨어**를 개발하기.***## 연구 방법저자들은 각 분류체계의 계층 구조(‘domain–phylum–class–order–family–genus–species’)를 컴퓨터 프로그램으로 분석해 서로 어떻게 연결될 수 있는지 ‘매핑(mapping)’이라는 절차로 비교했다.사용한 세 가지 비교 방법은 다음과 같다.- **Strict mapping(엄격 매핑)**: 같은 이름·같은 계급일 때만 일치로 인정.- **Loose mapping(느슨한 매핑)**: 같은 이름이면 계급이 약간 달라도 일치로 인정.- **Path comparison(경로 비교)**: 각 분류체계의 계층 구조(계통 경로)를 따라 유사도를 비교.이 분석 절차는 오픈소스 프로그램 **CrossClassify**로 구현됐다.***## 주요 결과1. **크기와 계층 깊이 차이**- NCBI와 OTT가 가장 방대한 분류체계로, **종(species) 단계까지** 내려간다.- SILVA와 RDP는 **속(genus)** 단계까지만 분류하며 상대적으로 단순하다.- Greengenes는 가장 작은 규모로, 일부 구간이 오래전 이후 업데이트되지 않아 최신성이 떨어진다.2. **공통 속명(Genus)과 과(Family) 수준 비교**- 다섯 분류체계가 공유하는 이름은 매우 적었다.- 예를 들어 90% 가까운 속(genus) 이름이 특정 분류체계에만 존재했다.- **SILVA와 NCBI** 사이의 일치는 비교적 높았으며, **RDP와 Greengenes**는 일치율이 낮았다.3. **매핑 결과의 일관성**- SILVA, RDP, Greengenes는 NCBI로 옮기기가 비교적 수월했지만, 그 반대(즉 NCBI에서 다른 분류로)는 잘 되지 않았다.- 가장 큰 OTT 체계는 다섯 분류체계를 모두 잘 포함할 수 있었으며, NCBI와 OTT 간에는 높은 유사도가 나타났다.- 전체적으로 **SILVA, RDP, Greengenes → NCBI/OTT 방향**으로의 변환이 원활했다.4. **수학적 분석 결과**- 유사도를 0(완전히 같음)~1(완전히 다름)으로 나타냈을 때,느슨한 매핑에서 NCBI와 OTT의 평균 차이값은 약 0.08~0.06으로 매우 낮았고,Greengenes는 약 0.77로 가장 달랐다.- 즉, NCBI와 OTT는 거의 비슷한 구조를 가진 반면, Greengenes는 독자적이고 제한된 분류를 가진다.***## 고찰이 연구는 미생물 분류체계의 한계와 불일치를 구체적으로 보여준다.특히 **Greengenes**와 **RDP**처럼 특정 영역(박테리아 16S rRNA 분석용)에 특화된 체계는 다른 분류체계와 호환성이 떨어져, 연구 간 결과 비교에 어려움을 만든다.반면 **NCBI** 분류체계는 방대한 데이터베이스와 폭넓은 생물 종을 포함하기 때문에, 다른 체계로부터 쉽게 대응(mapping)된다.그러나 OTT(Open Tree of Life)는 구조적으로 이상적이지만, 아직 관련한 DNA 서열 데이터베이스가 부족해 실용성이 떨어진다.따라서 현재로서는 **NCBI 분류체계를 공통 기준으로 사용하는 것이 가장 적절하다**는 결론에 도달했다.***## 의의와 시사점이 연구의 가장 큰 의의는 **서로 다른 미생물 분류체계를 직접 연결할 수 있는 알고리즘을 제시했다**는 점이다. 이를 통해 연구자들은 서로 다른 데이터 분석 결과를 변환하거나 비교할 수 있다.또한 프로그램 **CrossClassify**를 공개해, 미생물유전체 연구에서 일관된 데이터 비교를 가능하게 했다.***## 이 연구가 중요한 이유현대 생명과학에서는 수많은 미생물 데이터를 분석하지만, 같은 DNA 데이터를 연구자마다 다른 분류체계로 해석한다면 결과는 호환되지 않는다.이 논문은 그러한 **‘언어의 불일치’를 해소하는 번역기 역할**을 한다.이를 통해 서로 다른 실험과 연구 간의 결과를 공통된 기준으로 비교할 수 있으며, 궁극적으로는 **지구상 미생물의 진화적 관계를 하나의 거대한 계통수로 통합하는 데 기여**한다.다시 말해, 미생물 분류의 통합은 각기 다른 연구를 잇는 ‘공용 언어’를 만드는 작업이며, 이 논문은 그 중요한 첫걸음을 제시했다.
 |
| 출처: @ye._.vely618 |